A pheochromocytoma is a rare tumor that originates from the adrenal glands, which are located on top of the kidneys. These tumors typically develop in the adrenal medulla, the inner part of the adrenal glands. They are neuroendocrine tumors that produce excessive amounts of catecholamines, such as adrenaline (epinephrine) and noradrenaline (norepinephrine).

Adrenal glands play a crucial role in the body’s stress response, secreting hormones like adrenaline and noradrenaline. These hormones regulate various bodily functions, including heart rate, blood pressure, and the body’s response to stress. In pheochromocytoma, the tumor cells produce an excessive amount of these hormones, leading to a surge in their levels in the bloodstream.
Genetics can play a role in pheochromocytoma, particularly in familial or hereditary cases. Some genetic syndromes, such as Multiple Endocrine Neoplasia Type 2 (MEN2) or Von Hippel-Lindau (VHL) syndrome, predispose individuals to developing these tumors. Inherited genetic mutations associated with these syndromes increase the likelihood of developing pheochromocytomas and other tumors.
These tumors can cause a range of symptoms due to the excess hormones, including high blood pressure (hypertension), palpitations, headaches, sweating, anxiety, and sometimes even severe complications like heart problems or strokes.
Symptoms
Pheochromocytoma presents a diverse array of symptoms, often stemming from the excessive production of adrenaline (epinephrine) and noradrenaline (norepinephrine) by the tumor cells in the adrenal glands. These symptoms can vary in intensity and may appear intermittently, making diagnosis challenging. Common symptoms include:
High Blood Pressure (Hypertension): Pheochromocytoma often causes severe hypertension that can be intermittent or persistent. Blood pressure spikes are a hallmark, leading to headaches, dizziness, or visual disturbances.
Palpitations and Rapid Heart Rate: Due to the increased release of adrenaline, individuals might experience a pounding or rapid heart rate (tachycardia), leading to a sensation of palpitations or a fluttering feeling in the chest.
Headaches: Intense and severe headaches, sometimes described as migraine-like, may occur, often accompanying episodes of high blood pressure.
Sweating and Flushing: Profuse sweating, particularly excessive sweating without exertion or heat, and episodes of flushing or reddening of the skin may be observed.
Anxiety or Panic Attacks: Feelings of anxiety, nervousness, or panic attacks can be triggered by the surge of adrenaline released by the tumor.
Tremors or Shakiness: Some individuals might experience tremors, shaking, or a sense of inner restlessness.
Abdominal Pain: Pain or discomfort in the abdomen, especially in the region of the adrenal glands, might be present but is not always a prominent symptom.
Weight Loss: Unintentional weight loss might occur due to increased metabolism caused by the excess catecholamines.
Other Symptoms: Other less common symptoms include nausea, vomiting, constipation, and in rare cases, chest pain or breathing difficulties.
It’s important to note that not everyone with a pheochromocytoma will experience all of these symptoms. Some individuals might have only a few symptoms, while others may exhibit a broader range. The episodic nature of these symptoms often leads to misdiagnosis or delayed diagnosis, making it crucial for individuals experiencing such symptoms, particularly intermittent high blood pressure, to seek medical evaluation for prompt diagnosis and appropriate management.
How is Pheochromocytoma Diagnosed?
Diagnosing pheochromocytoma involves a multifaceted approach that includes clinical assessments, laboratory tests, and imaging studies. Here’s a detailed overview of the diagnostic process:
Clinical Assessment: The diagnostic journey for pheochromocytoma begins with a meticulous clinical assessment. The medical history and physical examination help identify symptoms such as hypertension, palpitations, headaches, and sweating, guiding suspicions towards this rare adrenal tumor.
Hormone Tests: Crucial for confirmation, blood and urine tests play a pivotal role in measuring catecholamines and metanephrines. Elevated levels during symptomatic episodes provide strong indications of pheochromocytoma.
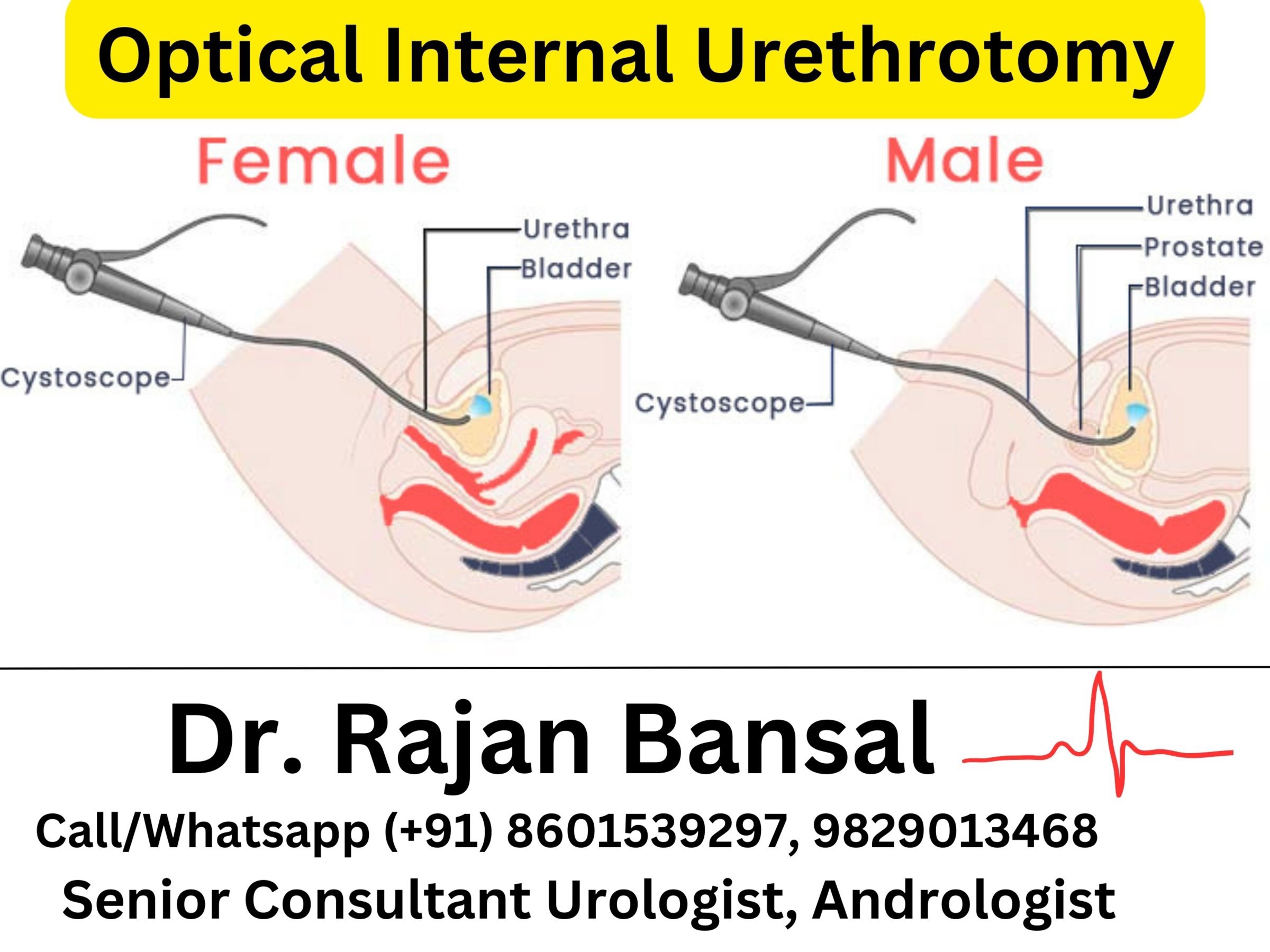
Imaging Studies: CT and MRI: Radiological imaging, particularly CT scans and MRI, forms the cornerstone of localization and characterization. Computed Tomography (CT) offers detailed cross-sectional images, precisely visualizing the adrenal glands and detecting tumors. Magnetic Resonance Imaging (MRI) provides high-resolution images, aiding in the assessment of tumor size, location, and adjacent tissue involvement.
Functional Imaging Techniques: In cases where conventional imaging falls short, functional imaging techniques like Metaiodobenzylguanidine (MIBG) scans or Positron Emission Tomography (PET) using specific radiolabeled tracers become invaluable. These studies enhance tumor visualization, capturing elusive tumors that may not be readily identified by CT or MRI alone.
Genetic Testing: For suspected hereditary forms, genetic testing explores familial predispositions, focusing on genetic mutations associated with syndromes like Multiple Endocrine Neoplasia Type 2 (MEN2) or Von Hippel-Lindau (VHL), aiding in accurate diagnoses and tailored management plans.
Provocative Testing: Sometimes, provocative tests, like clonidine suppression or glucagon stimulation, provoke a hormonal response, aiding in confirmation when clinical suspicion remains high.
A comprehensive diagnostic regimen encompassing clinical evaluations, hormone assays, advanced imaging modalities such as CT and MRI, functional studies, genetic insights, and occasionally provocative testing, forms the bedrock for the precise identification and localization of pheochromocytomas. This integrated approach ensures accurate diagnosis, guiding timely interventions and personalized treatment strategies for optimal patient outcomes.
Treatment of Pheochromocytoma at Institute of Urology, Jaipur
Treating pheochromocytomas involves a multidisciplinary approach that aims to manage symptoms, control hormone secretion, and ultimately remove the tumor. Here’s a comprehensive overview:
Preoperative Preparation:
Before surgical removal of the tumor (adrenalectomy), preoperative management focuses on controlling hypertension and normalizing blood pressure to minimize the risk of hypertensive crises during surgery. Medications called alpha-blockers or alpha-beta blockers are commonly used to stabilize blood pressure. Adequate hydration and electrolyte balance are also ensured before surgery.
Surgical Intervention – Adrenalectomy:
The primary treatment for pheochromocytoma is surgical removal of the tumor through adrenalectomy. This can be performed using open surgery or minimally invasive techniques such as laparoscopic or robotic-assisted surgery. The choice of approach depends on the tumor size, location, and the patient’s overall health. Minimally invasive surgeries offer quicker recovery times and reduced postoperative discomfort.
Medication Management:
In cases where surgical intervention is delayed (as per patient’s request) or contraindicated, medications are used to manage symptoms and control hormone secretion. These medications include alpha-blockers, beta-blockers, or calcium channel blockers to help control blood pressure and manage symptoms such as palpitations, headaches, and sweating. These drugs are usually used as a bridge therapy before surgery or for palliative management in unresectable cases.
Long-Term Follow-Up:
After successful surgical removal of the tumor, regular follow-up appointments are crucial to monitor for tumor recurrence, hormonal levels, and blood pressure control. Recurrence of pheochromocytoma is rare but possible, especially in cases of hereditary forms or incomplete tumor removal.
Hereditary Cases and Genetic Counseling:
In cases of familial pheochromocytoma syndromes, genetic counseling and screening for family members are recommended to identify individuals at risk and facilitate early detection and management.
Lifestyle Modifications:
Patients are often advised to maintain a healthy lifestyle, including regular exercise, a balanced diet, and stress management techniques. Monitoring blood pressure and adhering to prescribed medications are essential components of long-term management.
The treatment of pheochromocytoma revolves around surgical removal of the tumor, supported by preoperative management to stabilize blood pressure and postoperative follow-up for monitoring. In cases where surgery is not feasible, medications play a crucial role in symptom control and hormonal management. Overall, a comprehensive approach involving surgery, medication, and long-term monitoring ensures effective management and improved outcomes for individuals with pheochromocytoma.
Long Term Prognosis
In the majority of cases, the long-term prognosis for individuals with pheochromocytoma is excellent, especially after successful surgical removal of the tumor. Here’s an overview:
Surgical Cure: Surgical removal of the tumor through adrenalectomy is often curative, particularly for benign, localized pheochromocytomas. The majority of patients experience resolution of symptoms and normalization of blood pressure following successful surgery.
Low Recurrence Rate: After complete tumor removal, the recurrence rate of pheochromocytoma is low, especially in sporadic cases. Regular follow-up appointments are crucial for monitoring, but recurrence is rare.
Hereditary Syndromes: In cases where pheochromocytoma is associated with hereditary syndromes such as Multiple Endocrine Neoplasia Type 2 (MEN2) or Von Hippel-Lindau (VHL) syndrome, the risk of developing multiple tumors or recurrence is higher. Lifelong monitoring and proactive management are essential for these individuals.
Quality of Life: Successful treatment, including surgery and/or medications, significantly improves symptoms and enhances the quality of life for individuals with pheochromocytoma. Relief from symptoms such as hypertension, palpitations, headaches, and anxiety contributes to an improved overall well-being.
Long-Term Monitoring: Regular follow-up appointments, including imaging studies and hormone tests, are necessary post-surgery to monitor for any signs of tumor recurrence or hormonal changes. Long-term monitoring ensures early detection and prompt intervention if any recurrence or new tumors occur.
Lifestyle Management: Adhering to a healthy lifestyle, including regular exercise, a balanced diet, stress management techniques, and compliance with prescribed medications, plays a crucial role in maintaining overall health and managing blood pressure in the long term.
Overall, with timely diagnosis, appropriate treatment, and diligent long-term monitoring, the long-term prognosis for individuals with pheochromocytoma is generally favorable. Close collaboration with healthcare providers, adherence to follow-up recommendations, and a proactive approach to managing this condition contribute to an improved prognosis and quality of life.
Pheochromocytoma Treatment at Institute of Urology, Jaipur
Pheochromocytoma, though rare, underscores the critical need for expert care from urologists and endocrine specialists. Early diagnosis and timely intervention under the guidance of these skilled professionals are paramount. Swift identification and treatment of this adrenal tumor significantly alleviate symptoms, prevent potentially severe complications, and improve long-term outcomes. Collaborative efforts between patients, healthcare providers, and specialized teams ensure tailored treatment approaches, emphasizing the importance of precision in surgical intervention and meticulous postoperative care. Vigilance in recognizing symptoms, coupled with prompt medical attention and follow-up, remains pivotal in mitigating risks, offering optimal management, and ultimately enhancing the quality of life for individuals grappling with this intricate neuroendocrine condition.
At Institute of Urology, we specialise in an integrated approach towards managing conditions like Pheochromocytoma. If you are dealing with Pheochromocytoma, do not be skeptical about it and come and see us today. You can book an appointment for visit to the hospital at 9829013468 beforehand so that we can attend to you without any wait time. Please bring all your medical reports when you come for consultation so that we can take the protocol further as soon as possible.
Online video consultation is available with prior consultation. You can discuss your query with Dr. Rajan Bansal ( 8601539297 ) or Dr. M. Roychowdhury ( 9929513468 ) before coming to the hospital from the comfort of your home. At Institute of Urology, Jaipur, we ensure complete confidentiality and priority care.